Familial Dysbetalipoproteinemia is characterized by accumulation of abnormal beta lipoproteins, which resemble normal pre-beta migrating VLDL in size and flotation characteristics, but differ from them in composition. Planar xanthomas, particularly in the palmar and digital creases, are peculiar to this form of primary hyperlipoproteinemia, but tuberous, tuberoeruptive, and tendinous lesions also occur. It is much rarer than familial hyperbetalipoproteinemia; in the older literature, it was frequently called xanthoma tuberous. The mode of inheritance is uncertain.
Etiology and Pathogenesis Of Familial Dysbetalipoproteinemia
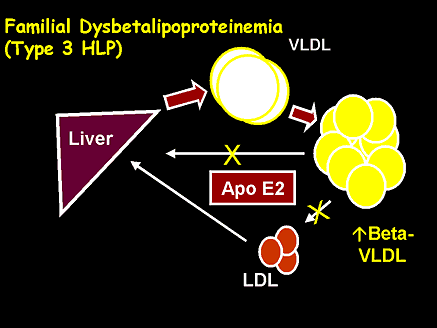
The abnormal lipoprotein contains predominantly the B apoprotein together with a basic protein, unusually rich in arginine, in particles with more cholesteryl ester and less triglyceride than normal VLDL. Its electrophoretic mobility is also ab. normal, encompassing the beta and extending toward the pre-beta region (“broad beta”). This lipoprotein is thought to represent remnant-VLDL which accumulate because of abnormality in the mechanism responsible for uptake of cholesteryl esters in the liver.
Clinical Manifestations of Familial dysbetalipoproteinemia
Chylomicronemia develops as soon as fat is ingested, and the disorder has been diag- nosed during the first week of life. Pancreatitis is very common, and many patients diagnosed in adult life have a history of bouts of abdominal pain in childhood. Abdominal pain, not definable as pancreatitis, has been reported frequently. but causes other than pancreatitis have not been established. Other complications, described rarely, include melena and pretibial ulcers. Like the pancreatitis, they may result from ischemia related to agglomeration of chylomicrons in capillaries. Hepa- tosplenomegaly, presumably the result of phagocytosis of chylomicrons by reticuloendothelial cells, is common, and hypersplenism has been described.
Foam cells may be found in the bone marrow. The endogenous hyperli- pemia occurring normally during pregnancy is greatly magnified and may lead to repeated attacks of pancreatitis. In infants and children especially, eruptive xanth- omas—srnall yellow papules in the skin, surrounded by an erythematous halo may occur when lipemia is severe.
Familial Dysbetalipoproteinemia Diagnosis.
Markedly lactescent blood plasma in a young patient with severe midabdominal pain and no history of alcoholism is presumptive evidence for this disorder or one of the other primary hyperlipemias. In exogenous hyperlipemia, the chylomicrons rise to form a creamy layer when the serum is allowed to stand overnight in the refrigerator and the infranatant serum is usually limpid. Triglyceride concentration is in- creased far more than that of cholesterol, and the chylomicron band predominates on electrophoresis.
Triglyceride levels fall dramatically when intake of dietary fat is severely restricted for two to three days. Lipolytic activity, measured in blood plasma obtained ten minutes after intravenous injection of 0.2 mg of heparin per kilogram of body weight, is reduced, and activity sensitive to inhibition by 0.5 M NaCl (a characteristic of lipoprotein lipase) is virtually undetectable.
Familial Dysbetalipoproteinemia Treatment.
The only established measure is restric- tion of dietary fat (“saturated” and “polyunsaturated” fats are equivalent) suffcient to reduce fasting triglyceride levels to 500 mg per 100 ml or less. This usually requires an intake of no more than grams per day by adults. On this regimen, xanthomas disappear and pancreatitis is uniformly prevented. Pretibial ulcers have also healed, and hypersplenism has subsided on this regimen. Once pancreatitis occurs, recurrence is very common when large amounts of fat are ingested and may happen within hours. During pregnancy, cornplete elimination of fat from the diet may be necessary to control hyperlipemia and minimize the possibility of pancreatitis.