Scleroderma is a disease involving blood vessels and connective tissue. The clinical picture is dominated by symptoms of vascular insufficiency caused by abnormalities in small arterioles and capillaries, and by progressive fibrosis in multiple organs. Most patients have involvement predominantly of the arms and hands with Raynaud’s phenomenon and acrosclerosis. The face and upper chest may be affected as well, but it is interesting that the legs and feet are involved less often, and it is rare that diffuse involvement of the entire skin is seen. Associated with the cutaneous manifestations are dysfunctions of certain viscera, particularly the esophagus, lungs, and kidneys.

The diagnosis of systemic sclerosis is made most often in patients between the ages of 35 and 55. Between three and five new cases per million population appear each year. Only 8 per cent of cases begin in the first two decades of life. The disease is three times more common in females than in males. There is some evidence that black women have a poorer prognosis than white women, and males a poorer prognosis than females.
Pathogenesis and Pathology of Scleroderma
Pathogenesis. Data are accumulating to indicate that abnormalities in the vascular system are primarily involved in the pathogenesis of this disease. Raynaud’s phenomenon with its characteristic blanching and pain followed by suffusion is a very common initial complaint in systemic sclerosis. Patients with Raynaud’s phenomenon have been shown to have significantly decreased cutaneous fingertip blood flow compared with that of normal controls when both were cooled to 18°C. In clinically unaffected muscle tissue, patients with scleroderma have been shown to have loss of 80 per cent of the normal amount of capillaries; those remaining capillaries have a diameter increased over normal, swollen endothelial cells, and reduplication of the basement membrane. Surface microvessels of the nail fold in systemic sclerosis are also decreased in number and increased in diameter.
Digital arteriograms in patients with Raynaud’s syndrome and scleroderma often have shown obstruction of the proper digital arteries, and plethysmography has demonstrated decreased amplitude of reflection of the pulse. There may be a common factor of altered vascular reactivity in the development of Raynaud’s phenomenon, depressed sensitivity to cholinergic agonists of the lower esophageal sphincter, pulmonary hypertension, and reduced renal cortical blood flow-all abnormalities associated with systemic sclerosis. Resting venous catecholamine concentrations and urinary catecholamine excretion patterns in scleroderma have been normal.
Studies of the proliferative lesion of scleroderma (the appearance of excessive and inappropriate collagen deposition) have revealed no significant abnormality of physical properties, amino acid analyses, cross-linking, or solubility of collagen. Detailed sequence studies have not been carried out in scleroderma collagen; it is not known whether the thin “beaded” filaments in collagen of scleroderma which resemble ”embryonic” fibrils actually do represent «1 (IIII collagen —a genetically different type of collagen « chains recently characterized from many soft tissues, including blood vessels and fetal skin.
Numerous studies have indicated (lint there is an increased rate of collagen synthesis in Hcleroderma Increased activity of protocollagen proline hydroxylase has been found in skin biopsies from scleroderma. Medium from cultures in vitro of .scleroderma skin fibroblasts has been found to contain significantly more collagen than does medium from control cells. A similar increase in the synthet ic rate of sialic acid, a proteoglycan component of connective tissue, has been found. No defect in the collagenolytic system in scleroderma has been reported.
To sum up: A leading yet unproved hypothesis for the development of systemic sclerosis is that a primary’ abnormality of the small blood vessels causes change in connective tissue sufficient to alter normal patterns of collagen and proteoglycan metabolism.
There is very little evidence that tissue damage in scleroderma is mediated by immune complexes or activa-t ion of the complement or kinin systems. Alt hough only rare reports have revealed gamma globulin deposited along renal basement membranes in scleroderma, fibrin as ii component of fibrinoid material in walls of small vessels of “scleroderma kidneys” is commonly found Pathology. The result of systemic involvement in scleroderma is sclerosis. The reticular dermis is usually thickened. It may have a normal collagen-hundle pattern or may show broad, homogeneous, acellular deposits of collagen with indistinct bundle patterns. Other findings include atrophy of the rete pegs of the epidermis, atrophy of the hair follicles and sweat glands, perivascular lymphocytic infiltration, and hyalinization of arterioles During acute, early phases of the disease, edema may be present in the dermis.
The subcutaneous tissue is replaced by thick collagen bundles which bind the dermis to deeper structures Pathologic changes in the musculoskeletal system include acute and chronic inflamma tion in the synovium with no pannus formation and with more sclerosis than is found 111 rheumatoid synovium with an equivalent inflammatory response. Fibrin deposits are laid down around tendons, and in muscles there are a variety of abnormalities, the most common living fibrosis of the perimysium and epimysium, scattered cellular infiltrates, and atrophy and necrosis of muscle fibers similar to that seen in polymyositis.
In the internal organs, microvascular abnormalities (see Pathogenesis), mild inflammation, and edema in connective tissue are followed by increased deposition of fibrous tissue in both appropriate and inappropriate loci. This leads to distortion of the architecture of the tissues affected. In the lungs, a relatively low-grade interstitial pneumonitis is followed by interstitial fibrosis, most marked in lower lobes. After this, cyst formation and bronchiectasis may develop. Arteriolar thickening (concentric intimal proliferation or medial hypertrophy) is seen, particularly in those patients with clinical evidence of pulmonary hypertension. In the gastrointestinal tract, the esophagus is frequently involved with muscle atrophy and fibrosis. Lesions secondary to reflux of gastric contents are present in 20 per cent. Fibrosis around Brunner’s glands in the submucosa of the duodenum occurs and can be recognized on specimens obtained by peroral biopsy. Involvement of the small bowel begins with patchy subserosal fibrosis and may progress to almost complete replacement of smooth muscle with fibrous tissue and marked thickening of the serosa. The small bowel may develop multiple saccula-tions, presumably at sites of weakness in the continuity of the wall. Dilation, muscle atrophy, and fibrosis are seen in the colon; the fibrosis is irregular, leading to the characteristic sacculations and diverticula.
The heart is frequently enlarged and may be the only organ weighing more than predicted for the subject’s body weight. Small patches of interstitial myocardial fibrosis are commonly found. In very severe cases, as much as 60 per cent of cardiac muscle is replaced by dense, relatively acellular and avascular fibrous tissue. Endocardial or valvular thickening is unusual and is rarely of hemodynamic significance Fibrinous pericarditis is found quite often, even in the absence of uremia. Kidneys in scleroderma are normal in size when renal involvement has not been present clinically. In patients dying with uremia they may be small and frequently have small cortical infarcts. Histologically, intimal proliferation in the interlobular arteries, fibrinoid necrosis of small arteries and arterioles (including the glomerular tufts), and thickening of the basement membrane (the “wire-loop” lesion) may all be present These changes are similar to those seen in kidneys from patients with malignant hypertension and occasionally may be present in the absence of renal failure or severe hypertension.
Clinical Manifestations and Diagnoses of Scleroderma.
The disease usually begins insidiously. Raynaud’s phenomenon, vague weakness, weight loss, diffuse stiffness and aching, polyarticular arthritis, and diffuse edema of the hands are the most common initial symptoms. If the illness progresses, it generally does so slowly, and is marked by characteristic organ system changes. Rarely, rapid progression to severe cutaneous and visceral involvement may occur in less than six months.
Cutaneous System.
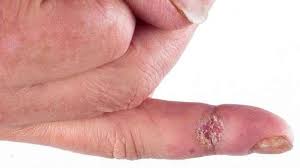
The patient with classic, well-developed acrosclerosis presents with taut, thickened, or edematous skin bound tightly to subcutaneous tissues in the hands and fingers. Feet and toes are involved less often than hands, forearms, and neck. Normal skin folds at the knuckles disappear. Chronic recurrent painful ulcerations at the ends of the digits develop, and the fingers themselves may shorten through progressive resorption of the terminal phalanges. Joints become immobilized from tight encasement in thickened skin as well as jrom contractures of muscles and tendons and palmar fascia. Telangiectasia, increased or decreased pigmentation, and subcutaneous calcification are common Hair becomes thin, and the skin of the face appears smooth and waxy. The skin around the mouth may constrict, restricting lip movement and preventing adequate dental hygiene. The normal sweating mechanism is often impaired; the involved skin feels leathery and dry and may scale and itch. The CRST syndrome (calcinosis. Raynaud’s phenomenon, sclerodactyly, and telangiectasia) and the Thibierge Weissenbach syndrome idiffuse deposition of insoluble calcium phosphates in subcutaneous tissue in the presence of acrosclerosisi are variants of the cutaneous expression of systemic sclerosis.
Almost half the patients with systemic sclerosis present with joint pain or develop it during the first year of their illness Small joints are involved more often than lurgeones Joint deformity and immobility in systemic sclerosis do not result from an invasive. erosive synovitis as in rheumatoid arthritis, but are the result of encasement as subcutaneous tissue is replaced by bundles of collagen. Muscle wasting is often severe in areas such us the hands, in which joint mobility is impaired by progressive tightening of the overlying skin.
Gastrointestinal Tract. Of the internal organ systems, the gastrointestinal tract is the one most often involved in systemic sclerosis. Oral symptoms include xerostomia and a progressive decrease in the size of the mouth. Sjttgren’s syndrome is seen often, and the frequency of its association with systemic sclerosis is probably underestimated Symptoms referable to the esophagus, ranging from simple dysphagia to heartburn, nausea, and substernal fullness, are found in 45 to 60 per cent of cases. The dysphagia is related to diminished peristalsis in the lower segments of the esophagus and incompetence of the lower esophageal sphincter If reflex esophagitis becomes a persistent complication, stricture may develop.
There is a very frequent association of Raynaud’s phenomenon and decreased esophageal peristalsis. although this association is not limited to patients with scleroderma. Vomiting, abdominal distention. und puin or diarrhea may indicate involvement of the small intestine As in the esophagus, motility of the small bowel is decreased, and there may be malabsorption secondary to intraluminal stagnation with cun-comitunt bacterial overgrowth. Functional bowel com-pluints secondary to pathologic changes in the colon are common. Disease of both large and small bowel may produce a clinical picture identical to paralytic ileus with incomplete obstruction at any level. Pneumatosis cystoides intestinal is uir-filled cysts in the mesentery which may rupture, causing peritonitis) is a rare but striking complication of scleroderma bowel disease.
Differential Diagnosis of Scleroderma;
When systemic sclerosis presents as persistent symmetrical polyarthritis involving the hands without previous skin changes, it may be impossible to differentiate it from rheumatoid arthritis or systemic lupus erythematosus, or, if the overlying skin becomes edematous or red, dermatomyositis. In fact, “overlap” syndromes combining clinical or serologic manifestations of several of these entities are being described with increasing frequency, and one should avoid hasty classification of any clinical picture that is consistent with more than one of the connective tissue diseases
Diseases or pathologic findings associated with Raynaud’s phenomenon which must be differentiated from scleroderma include occupational trauma; anatomic lesions. e.g.. scalenus anticus syndrome and cervical ribs; vasomotor syndrome’ resulting in disuse atrophy, e.g.. Sudek’s atrophy or the shoulder-hand syndrome; peripheral vascular arteriosclerosis; heavy metal or ergot poisoning. and hematologic abnormalities, e.g., polycythemia vera. paroxysmal cold hemoglobinuria, and presence of cold agglutinins or cryoproteinemia.
The atrophic changes in the skin in systemic sclerosis must be differentiated from those seen in Werner’s syndrome, progeria. Hutchinson-Gilford syndrome), chronic hypostatic edema or myxedema, lichen sclerosus et atro-phicus. porphyria cutanea tarda, and/or scleredema. Unlike systemic sclerosis. Werner’s syndrome affects the feet more severely than the hands, and has associated with it a high-pitched voice, growth abnormalities, and premature cataracts, arteriosclerosis, and diabetes Sclerodermatous changes have been reported in skin of patients with progcriu. but the dwarf-like stature, characteristic facies, and premature death from coronary insufficiency M-t this syndrome apart. Porphyria cutanea tarda may have skin changes simulating scleroderma but can Ik* difTcrcntiaU’d on the basis of urinary or fecal porphyrin excretion Scleredema (scleredema adultorum of Huschko is a brawny edema which appears abruptly, involves skin of the neck and chest initially, and is characterized pathologically by the presence of a material resembling acid mucopolysaccharide diffusely interspersed among collagen bundles in the dermis. This is a self-limited process, perhaps related to a previous bacterial infection, and has a good prognosis. The cutaneous telangiectasia of scleroderma resembles Osier-‘Weber-Rendu disease hereditary telangiectasia).
Involvement of viscera in systemic sclerosis can mimic many other illnesses. The abnormalities in pulmonary function and the roentgenographic findings seen are similar to those found in idiopathic pulmonary fibrosis (Mamman-Kich syndrome) and the pulmonary fibrosis of rheumatoid arthritis or advanced sarcoidosis. Scleroderma heart disease must be differentiated from infiltrative cardiomyopathies as well as fibrosis from diffuse cor onary artery disease The dysphagia of scleroderma is completely nonspecific, and disease of the bowel rarely can simulate sprue or any of the malabsorption syndromes, partial intestinal obstruction, or megacolon.
Treatment of Scleroderma
Patients with acrosclerosis can lead productive and useful lives. The most important goal in therapy is to preserve function in and prevent injury to the hands. Vocational and/or climatic change may be indicated Hand care must Ik* stressed, including instructions for active and passive exercises that the patient may do himself several times each day in order to prevent further deformity. Early signs of local infection in fingertips must be treated immediate!) In-fore they progress to large ulcerations. Psychologic support can be of great value in helping a patient adjust to th«- discomfort and apparently inexorable progression of his disease
Because so little is known about the underlying defect in scleroderma or its pathogenesis, no specific treatment is available. The one possible exception to this is the use of broad-spectrum antimicrobials in treatment of malabsorption associated with bacterial overgrowth in the upper small intestine. If esophageal motility is decreased. antacid therapy is used liberally to prevent secondary esophagitis and stricture. If stricture develops, bougienage will be needed. Salicylates in pharmacologic doses may decrease clinical signs of inflammation, e.g., elevated sedimentation rates, joint pain, and erythema, which is present in most patients at somt* time in the course of their disease.
Corticosteroids are ineffective in the treatment of sys-temic sclerosis; neither the vascular lemons nor the fibrosis are improved by their use However, there is a subgroup of patients with an overlap syndrome called mixed connective tissue disease” who present with a variety of symptoms, including Raynaud’s phenomenon, arthritis, and skin changes consistent with scleroderma, but with features (including LE cells, leukopenia, inflammatory myositis, and lymph adenopathy) of other connective tissue diseases as well. Many of this group will have high titers of antinuclear antibody in a “speckled” pattern directed against an extractable nuclear antigen ‘not DNA>. In general, this group of patients does not develop renal disease and responds very well to corticosteroid therapy.
Recently, use of intra-urterial reserpine has been advocated to decrease the frequency of Raynaud – phenomenon and potentiate healing of ulcers Given intru-ur-terially (0.5 to 1,0 mg), benefit without side effects has been noted up to seven months afler a single injection. A decrease in finger vasoconstriction in response to cooling in patients with scleroderma given reserpine in the ipsilateral brachial artery has been documented The placebo effect of this type of procedure and potential hazards of inlra-arterial reserpine must be continually eval-uated. Oral reserpine in doses sufficient to relieve symptoms of Raynaud’s phenomenon often causes central depression, but when used in small doses 0.5 mg per day), combined with oral sympathetic or ganglionic blocking agents, may increase capillary blood flow without causing intolerable side effects.
Potassium p-aminobenzoate, ethylenediaminetetra-acetic acid, 6-aminocaproic acid infusions, and dimethyl sulfoxide applications have been employed by a number of investigators D-penicillamine, capable of inhibiting formation of cross-links in collagen and some mammalian collagenases in vitro, has not heen of proved benefit in scleroderma Pilot studies of immunosuppressive therapy are being evaluated; initial results show no clear-cut role for these drugs in systemic sclerosis
Initial results in few cases in which “scleroderma kidneys” were removed and hemodialysis and/or renal transplantation carried out have demonstrated that life of good quality can he prolonged by these aggressive approaches. It is not yet known whether the transplanted kidneys will develop vascular changes, but it is now clear thut patients with systemic sclerosis who develop renal disease need not die from rapidly progressive renal failure.
TREATMENT OF SCLERODERMA
There is no treatment that leads to the cure of scleroderma.Already the systemic sclerosis is a disease of slow evolution, but permanent. The treatment aims to relieve symptoms and reduce the activity of the disease.
The drugs used depend on the symptoms that the patient has, for example:
- Vasodilators, such as nifedipine, are commonly used to treat Raynaud’s phenomenon.
- Proton pump inhibitors such as omeprazole, help in gastric and reflux symptoms.
- Anti-inflammatories and corticosteroids in low doses help in pain and stiffness of the joints.
- Immunosuppressive medications such as methotrexate or mycophenolate mofetil, help reduce damage to the skin and organs caused by the immune system.
- Antihypertensives to treat hypertension.
There is no treatment that is universal for all types of scleroderma. Each treatment must be established individually, according to the symptoms and the type of commitment of the existing organ in each patient.