Myasthenia gravis is usually defined as a state of abnormal fatigability. This definition has the merit of being consistent with the characteristic electrophysiologic disorder found in patients with this condition, but there are serious disadvantages. “Normal” fatigue is not defined, muscles weakened from any cause are apt to fatigue more readily than normal, and myasthenic muscle may be completely paralyzed (especially ocular muscles), with no signs of fatigue.
Moreover, emphasis on fatigue has led to the treatment of many psychoneurotic patients with cholinergic drugs. An alternative definition has therefore been proposed: “Myasthenia gravis is a disease characterized by weakness that has special characteristics: predilection for ocular it: ether cranial muscles; tendency to fluctuate daring brief periods of observation (in the course of a single day) or for longer periods remiss ions and exacerbations); with ho signs of neural lesion, and partial reversibility by the administration of cholinergic drugs.
Etiology.
The cause of myasthenia gravis is not known. Several disorders tend to occur more frequently in patients with myasthenia gravis, such as hyperthyroidism or other thyroidal disorders, and a significant number of patients with myasthenia gravis have thymomas. But in neither of these situations,, nor in others, has a causal relationship been proved. Whether or not there is a significant association with “autoimmune” diseases remains to be proved; rheumatoid arthritis and systemic lupus erythematosus have been reported in patients with myasthenia gravis, but the statistical significance of these associations is uncertain.
Several lines of evidence suggest an immunologic disorder in myasthenia gravis. Thymomas occur in about 15 per cent of all patients, more often in patients older than 40 years. Germinal centers are seen frequently in the thymus glands of patients who have no tumor. Infiltration of muscle by monocytes is characteristic, especially in perivascular collections called lymphorrhages
Antibodies to muscle can be demonstrated by immunofluorescent techniques in approximately a third of all patients with myasthenia, and in almost all patients with concomitant myasthenia and thymoma. However, the nature of the antigen that elicits these antibodies is uncertain, and it seems quite clear that the antibodies are not responsible for the symptoms of myasthenia. Thus, antibodies may be present in patients with thymoma who have no evidence of myasthenia. There is no correlation between the presence of antibodies in serum and the appearance of transient neonatal myasthenia, and the concentration of serum antibodies does not correlate with the severity of symptoms. Finally, physiologic evidence indicates a disorder of neuromuscular transmission in myasthenia, and the antibodies react with proteins in the myofiber, but not at the neuromuscular junction. The significance of these human antibodies is therefore uncertain. Moreover, no one has yet succeeded in designing a reproducible experimental model of the disease by immunologic techniques or by any other method.
Neuromuscular Disorder.
The symptoms of myasthenia gravis resemble those of curare intoxication. This observation led to the use of curare antagonists in the treatment of myasthenia gravis. Ail effective therapies have been derived from this initial observation, and all drugs currently in use are inhibitors of the enzyme cholinesterase. Thege drugs also partially repair a physiologic defect that can be detected in patients by relatively simple techniques- When the ulnar nerve of normal individuals is stimulated at rates of 25 per second or less, the action potential of the hypo- thenar muscles is sustained at a constant amplitude.
In patients with myasthenia, however, there is a rapid decline in the height of the evoked potentials. If the patient is then given neostigmine, the amplitude of the evoked potentials is restored to normal. This, pattern could be consistent with either excessive cholinesterase activity, inadequate release of acetylcholine, or blockade of the junction by some curare-like compound. Microelectrode studies have been performed upon intercostal muscle fibers of patients with myasthenia (after excision of these muscles, they can be studied in vitro with the most advanced techniques). Investigations of this nature suggest that the basic defect is a reduced amount of acetylcholine released in each quantum from the motor nerve terminal. This could be caused by either some defect in the binding of acetylcholine or the presence of a “false transmitter,” but how the defect comes about is not known.
Incidence and Prevalence.
The prevalence of myasthenia gravis is about 33 per million population and the annual incidence about 2 to 5 per million. Translated into the experience of a busy 1000-bed general hospital, about 20 new cases are encountered each year, or about the same incidence as for lupus erythematosus. Cases ocgur in aM decades of life, most frequently at about age 40. Among young adults, the disease affects women about three times more frequently than man. Among children and older persons, however, there is no sexual predilection.
Pathology.
Pathological changes in myasthenia gravis are limited to muscle and thymus. Skeletal muscles may appear completely normal, but there are often collections of lymphocytes around blood vessels. In some cases there is evidence of degeneration of muscle fibers, and the inflammatory cellular’ response may be more extensive. Rarely, similar lesions are encountered in the myocardium. The thymus gland is often abnormal. Encapsulated tumors (thymoma) occur in about 15 per cent of the cases, almost all after age 30. In about 25 per cent of these tumors there is evidence of local invasion, but distant metastases are virtually unknown in patients with myasthenia, and the local invasiveness is not a cause of symptoms, Lymphocytic proliferation, in the form of germinal centers, is seen in the thymus glands of almost all other patients, but. sometimes the gland appears normal, and in some older individuals the gland is so convoluted that it cannot be found.
Patients dying of this disease usually have some pulmonary – disorder (edema, atelectasis, infection) but this is secondary to the terminal events. Patients with myasthenia may live long enough to develop a carcinoma of any organ, but it has not been shown that myasthenia occurs more frequently in patients with cancer than might be expected on the basis of chance.
Clinical Manifestation of Myasthenia gravis
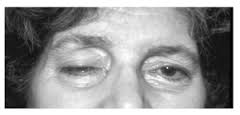
The symptoms of myasthenia gravis are all due to weakness. The most common presenting symptoms relate to weakness of eye muscles, ptosis, or diplopia. At the onset these symptoms may last only a few days, then disappear, only to return weeks or months later. Ptosis frequently varies even in the course of a single day. Diplopia may be noted in particular directions of gaze. Difficulty in chewing, dysarthria, and dysphagia are other common initial symptoms. Limb weakness is perhaps more often proximal in accentuation, so that patients have difficulty climbing stairs or rising from chairs, or have difficulty lifting heavy objects or raising the arms overhead.
However, distal weakness is not uncommon, and the initial symptoms may be related to the strength of the hands or fingers. Selective respiratory weakness is most unusual. There is no alteration of consciousness, no pain, and in untreated patients, no cramps or muscular twitching. Difficulty in eating and swallowing may lead to considerable loss of weight, but there is no specific wasting of muscle except in patients with severe chronic weakness.
On examination, evidence of weakness of the appropriate muscle groups is found.
If any sensory abnormality is found, there must be some other disorder, alone or in combination with myasthenia. Similarly, hyperactive reflexes and Babinski signs imply some other disorder, as does the complete loss of reflexes. Weakness is responsible for all the signs of myasthenia. Ptosis may be unilateral or bilateral. The patient may experience diplopia even when there is no obvious weakness of the ocular muscles; more often, there is weakness of several muscles in a pattern that cannot be explained by disorder of a particular ocular motor nerve and is not usually symmetrical in the two eyes.
In addition, there is frequently weakness of eye closure because of paresis of the orbicularis oculi. Muscles of the lower face are involved later. Lingual and palatal weakness are evident in patients with dysarthria. A nasal twang and indistinct speech imply that dysarthria has facial, lingual, palatal, and, sometimes, respiratory components.
In advanced cases, patients support the chin with one hand to help them talk, a maneuver that seems almost pathognomonic of myasthenia. Neck weakness may be mild or severe enough to cause difficulty in holding the head erect.
Limb weakness may be mild or so severe that the patient cannot walk or even turn in bed. Respiratory weakness does not occur alone; ventilatory insufficiency occurs only in severely affected patients and usually in generalized disease, but some patients with oropharyngeal weakness may be unable to breathe Unassisted even though limb muscles are strong. Even though weakness fluctuates, a normal neurologic examination is inconsistent with the diagnosis of myasthenia gravis if the patient is symptomatic at the time.
The Basics of Myasthenia Gravis Diagnosis.
In almost all cases the diagnosis of myasthenia is obvious from the history and examination. TKe diagnosis is immediately confirmed by the response to cholinergic drugs. For adults, 1 ml. of edrophonium (10 mg.) is given intermittently; after injection of 2 to 3 mg., specific muscle groups are evaluated. If there is no response within 30 seconds, the injection is continued to a total of 6 mg. and the patient is evaluated once again. If there is still no response, the remainder is given, a total of 10 mg. This drug is preferred when cranial muscles are being tested because the response is prompt and dramatic.
Cranial weakness cannot be simulated voluntarily, ar.d placebo injections are therefore not necessary If it is desired to evaluate limb strength, the injection of neostigmine has advantages because the effect lasts longer, permitting more leisurely testing. The adult dose is 1.5 mg. intramuscularly, and it is usually combined with atropine, 0.5 mg., to avoid the muscarinic symptoms of abdominal cramps and sweating. When limb strength is evaluated, it is sometimes advisable to evaluate placebo responses by giving the atropine 30 minutes before the neostigmine. In all cases of myasthenia gravis there is some response to these drugs, but the response is sometimes slight, and the test may have to be repeated on several occasions to provide convincing evidence.
To provide confirmatory evidence in some cases, the electromyographic response to ulnar nerve stimulation may be studied. The characteristic decline in amplitude of the evoked potential is usually seen in affected muscles, but the response may be normal when the disease is restricted to the eyes. Rarely, it is desirable to administer d- tubocurarine, a drug to which myasthenics are unusually sensitive. There are hazards in the administration of this drug, however, and it should probably be left to research centers; d-tubocura- rine should never be given without appropriate precautions to support respiration.
No other laboratory tests provide diagnostic information, but it is useful to perform a standard series of studies. Lamina Grams of the mediastinum are necessary to identify thymomas. Thyroid function should be tested in all myasthenics, and it is useful to evaluate immunologic disorders by serum protein electrophoresis, LE cell preparation,latex fixation test for rheumatoid factor, and anti- nuclear antibodies. Tests for muscle antibodies are performed in special centers.
The differential diagnosis of myasthenia includes most of neurology, but a few conditions require special consideration. Probably the most frequent error in diagnosis concerns patients with emotional fatigue states and hysterical weakness. There are no cranial muscle symptoms in these patients (except globus hystericus i, whereas cranial muscles are involved in virtually all myasthenics. Their symptoms are not those of weakness but of exhaustion, and in tests of strength against resistance there is apt to be marked variation in effort, or dramatic giving way.
Amyotrophic lateral sclerosis and peripheral neuropathy may be confused when these disorders afreet cranial muscles, but signs indicative of a neurogenic disorder distinguish them from myasthenia.
Polymyositis may be confusing, but ocular muscle paresis is not found. There is some debate about the specificity of the therapeutic effect of cholinergic drugs, but for practical purposes it may be taken that an authentic (not placebo), convincing (not equivocal), and reproducible (not seen by one examiner only) effect is found only in myasthenia .
Myasthenia Gravis Treatment.
Drug Therapy. The major drugs used to treat myasthenia gravis are inhibitors of cholinesterase, neostigmine, and pyridostigmine. Another drug, ambenonium. is also used in some clinics, but has no special advantages. It is best to use only one drug; there is no benefit from combinations of two or more. Neostigmine is provided in 15 mg. tablets, and pyridostigmine in 60 mg. tablets; these are essentially interchangeable. The choice of drug is arbitrary because there is no evidence that the maximal benefit achieved by one is more than that of the other. Most patients, ^ but not all, prefer pyridostigmine because it is less apt to cause abdominal cramps and diarrhea, and it is less apt to cause noticeable peaks and valleys of strength. But these advantages have never been proved objectively, and some patients do very well with neostigmine.
Pyridostigmine is also provided in a slow-release capsule (Timespan) of 180 mg., said to provide 60 mg. immediately and the remainder in 8 to 12 hours. Some clinicians use the prolonged-action preparation throughout the day, but because of uncertainties of release, it seems advisable to use this only at bedtime for patients who would otherwise have to awaken at night or who are very weak on waking in the morning. (For patients who cannot swallow pills, both drugs may be administered parenterally-, and pyridostigmine may be given as a syrup. The intramuscular dose is about one tenth of the oral dose, and the intravenous dose is one thirtieth of the oral dose.) Decisions about optimal dosage are sometimes difficult.
Some authorities advocate the use of edrophonium to evaluate oral drug therapy; a test dose of 2 mg. is given intravenously. If the patient improves, oral therapy has been too little. If there is aggravation of weakness, oral therapy has been too much.
If there is no change,oral therapy has been just right. But other authorities find this test an unreliable guide. If edrophonium is not used, there is nothing but clinical observation to guide the therapist. For mild cases, two tablets of either neostigmine or pyridostigmine three times daily, with meals, is useful. If there is inadequate response, the dose may be increased, first by shortening the interval between doses, then by increasing the quantity of drug with each dose. Problems arise because these drugs never completely reverse symptoms. Therefore, the dose should be increased only so long as there is clear-cut response, and it should not be increased beyond the amount giving some perceptible benefit.
Myasthenia Gravis Surgical Treatment.
Thymectomy is followed by significant improvement in about two-thirds of patients with myasthenia and no tumor. Patients with thymoma fare worse whether the tumor is excised or not. Chest surgeons, anesthesiologists, and respirator units for postoperative care are now so skillful that the risks of operation are almost negligible, and recent attempts to remove the gland through an incision in the neck rather than by splitting the sternum should reduce the morbidity considerably. Because not all patients improve, and because it is difficult to predict which patients will benefit, there is still some uncertainty in the selection of patients.
Some recommend the operation for all patients who are incapacitated in daily living for more than six months despite adequate drug therapy. In preparing patients for operation their usual oral dosage is continued until the day of operation, then stopped. Tracheostomy is performed for all patients with oropharyngeal weakness, and intermittent positive pressure breathing is used in the postoperative period. Specific drug therapy is withheld for several days, until fever subsides, and then started again at a level of one half or less of the preoperative dose.