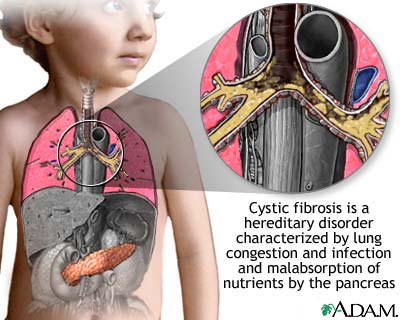
The Cystic fibrosis is a genetic disease caused by alterations in a gene called CFTR ( cystic fibrosis transmembrane regulator ), which creates an imbalance in the concentration of chloride and sodium in the cells that produce the body secretions, such as mucus and sweat (exocrine glands).

Prevalence of Cystic fibrosis.
Cystic fibrosis is transmitted as an autosomal recessive trait. The carrier rate of the abnormal gene is about 5 per cent in Caucasians, resulting in a prevalence of the homozygous state of 1 per 1,600 to 2,000 Caucasian (compared to 1 per 17,000 black) births. Homozygotes for the CF gene have almost all of the clinical features of the disease, but heterozygotes are clinically normal. The disease is recognized in most patients prior to adolescence; rarely the diagnosis may not be made until the third or fourth decade of life.
PATHOGENS of Cystic fibrosis.
The unknown primary biologic defect presumably leads secondarily to the changes in the physical characteristics of mucus in cystic fibrosis, which in turn lead to obstruction and dilatation of glands and their ducts. No primary abnormalities of the mucoproteins secreted, of ciliary function, or of host defenses (including cell-mediated, humoral, and secretory immunity) have been demonstrated. Thus, the reason for the obstruction of glands caused by abnormal mucous clearance remains unknown.
Symptoms of Cystic Fibrosis
The signs and symptoms of cystic fibrosis vary according to the age of the patient. In newborns, it is possible to observe:
- Bowel obstruction
- Difficulty gaining weight
- Cough with secretion
- Dehydration for no apparent reason.
- difficulty of growth, weight gain below normal, vitamin deficiency and malnutrition;
- abnormally bulky, greasy and foul-smelling stools;
- pneumonia and bronchitis;
- chronic sinusitis;
- constant coughs with phlegm and eventually blood;
- skin with salty taste, due to excess salt in sweat;
- appearance of nasal polyps (inflamed tissue inside the nose.
Over the years, the patient may present:
- Weight loss
- Progressive malnutrition
- Chronic cough with a lot of secretion
- Chronic sinusitis
- Formation of nasal polyps
- Liver disease ( biliary cirrhosis )
- Diabetes
- Respiratory infections
- Infertility
Cystic fibrosis DIAGNOSIS.
Cystic fibrosis is diagnosed in nearly 80 per cent of patients in the first three years of life. However, the diagnosis should be suspected in any adolescent or young adult presenting with chronic bronchopulmonary disease and pancreatic insufficiency. For diagnosing CF, a quantitative pilocarpine iontophoresis sweat test should be performed in a laboratory with established expertise. At least 50 mg of sweat should be collected and the results should be confirmed with a second test. Elevated sweat sodium and chloride concentrations, exceeding 60 mEq per liter in children and 70 mEq per liter in adults, are diagnostic of CF.
No screening tests are available for detecting the heterozygous state. Prenatal tests are available for the homozygous condition, but their reliability has not yet been established. In the first four to six weeks of life, when it is difficult to obtain sufficient sweat for testing, high serum concentrations of immunoreactive trypsin may prove to be diagnostic and can be used for routine newborn screening.
Other tests that identify problems that may be related to cystic fibrosis include:
- X-ray or chest computed tomography
- Stool fat test
- Lung function test
- Measurement of pancreatic function
- Secretin stimulation test
- Trypsin and chymotrypsin in feces
- Upper gastrointestinal tract (GI) and small intestine series
The earlier you discover cystic fibrosis, the better the quality of life and the longer the patient will live – since there is no cure for the disease.
TREATMENT of Cystic fibrosis.
An increasingly large number of patients with cystic fibrosis survive into adulthood, and an additional small number of patients with previously undiagnosed cystic fibrosis will be identified in young adulthood. The major source of morbidity in later life is pulmonary disease. Vigorous pulmonary toilet is basic, and good bronchial clearance can be obtained with a combination of chest physical therapy, active coughing, and exercise. Patients should be encouraged to pursue any form of exercise within their capacity. About 50 per cent of adult patients have bronchial hyper-reactivity with significant variability of air flow rates. Aerosols of bronchodi-lators in normal saline are sometimes partially effective. In occasional cases, air flow can be improved by oral corticosteroids without any evidence of worsening of the bronchopulmonary infection.
Antimicrobial agents are particularly important in the therapy of CF lung disease. Chronic antistaphylococcal therapy can be given orally for years without evidence of drug resistance or complications. Antipseudomonal agents are essential for acute exacerbations of bronchial infections and intermittent bouts of pneumonia. These infections generally remain localized in the lungs; bacteremia, empyema, and extrapulmonary spread are unusual. The initial treatment of Pseudomonas aeruginosa in adolescents and adults is most commonly with the combination of tobramycin and ticarcilbn. Subsequent modification should be based on the results of sputum cultures and in vitro sensitivity tests. When the acute or the chronic bronchopulmonary exacerbation is due to Pseudomonas cepacia, the antimicrobial choice is usually extremely limited.
Most patients with pancreatic insufficiency can be maintained on a normal diet with supplemental pancreatic enzymes The preferred agents are entenc-coated microsphere pancreatic enzyme preparations, such as PancTease and Cotazym-S, given in doses of three to five capsules per meal. The concomitant use of sodium bicarbonate or H:-receptor antagonists (cimeti-dine, ranitidine) may also improve the fat and protein absorption, since the efficacy of pancreatic enzymes is markedly increased at higher intraluminal pH levels.
“Meconium ileus equivalent” can usually be treated with enemas or nasogastric suction. Surgery is necessary on rare occasions. The administration of substantial amounts of pancreatic enzymes may reduce the frequency of this complication of intestinal obstruction. The lithogenicity of the bile can be reversed with appropriate pancreatic enzyme replacement. No treatment is available for the hepatic or vas deferens abnormalities. Nocturnal oxygen therapy is being evaluated to determine if prevention of sleeping artenal oxygen desaturation decreases cardiopulmonary morbidity and prolongs survival. The psychosocial aspects of this disease are formidable, and where possible patients should be cared for by special units so that their manifold problems can be dealt with by staff experienced in their management.
PROGNOSIS of Cystic fibrosis
About half of the patients with cystic fibrosis in the United States live to be 20 years of age. However, in areas where patients are cared for by special units, such as those supported by the Cystic Fibrosis Foundation and \’IH, 50 per cent may survive to at least 25 years of age, with males doing much better than females.
Factors associated with a better prognosis in cystic fibrosis are the male sex, maintenance of appropriate weight, single system (gastrointestinal or pulmonary) involvement at presentation. and a normal chest radiograph within the first year of presentation. Additional good prognostic factors are likely to be the presence of pancreatic function and the absence of Pseudomonas cepacia. Early diagnosis of cystic fibrosis has not been shown to influence the long-term survival of these patients.